Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by pathological T cell-B cell interactions. Expansion of T follicular helper (TFH) and T peripheral helper (TPH) cells, two types of T cells that provide help to B cells, is a prominent feature of SLE. Human TFH and TPH cells produce high levels of the B cell chemoattractant CXCL13. However, the regulation of T cell CXCL13 production and the relationship between CXCL13-producing T cells and other T cell states remains unclear.
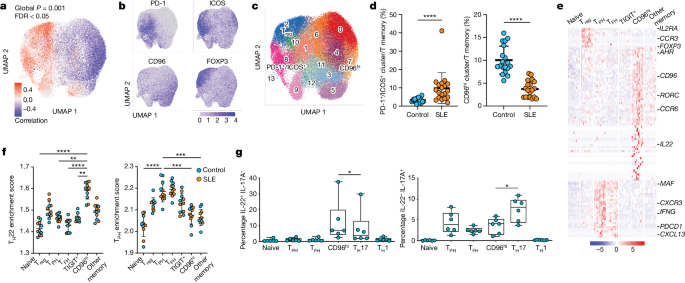
Recent studies have identified an imbalance in CD4+ T cell phenotypes in patients with SLE. Specifically, there is expansion of PD-1/ICOS CXCL13+ T cells and reduction of CD96hi IL-22+ T cells. Using CRISPR screens, researchers have identified the aryl hydrocarbon receptor (AHR) as a negative regulator of CXCL13 production by human CD4+ T cells. AHR coordinates with AP-1 family member JUN to prevent CXCL13+ TPH/TFH cell differentiation and promote an IL-22+ phenotype. Type I interferon opposes AHR and JUN to promote T cell production of CXCL13.
Scientists at Northwestern Medicine and Brigham and Women's Hospital have discovered molecular defects that promote the pathologic immune response in SLE. Insufficient activation of the AHR pathway results in an excess of disease-causing immune cells called T peripheral helper cells, which produce disease-causing autoantibodies. Activating the AHR pathway or limiting excessive interferon in the blood can reduce the number of these disease-causing cells, potentially curing lupus.
These findings provide new insights into the regulation of T cell CXCL13 production and suggest potential therapeutic strategies for treating SLE. However, it is important to note that more research is needed to fully understand the complex interplay between different T cell populations and their roles in autoimmune diseases like SLE.